
Mitogen Binding, Cytoskeleton Patterns and Motility of T
Lymphocytes in Microgravity
M. Cogoli-Greuter, B. Bechler, Lorenzi & A. Cogoli
Space Biology Group, ETH Technopark, CH-8005 Zürich,
Switzerland.
L. Sciola, P. Pippia, G. Sechi
Institute of General Physiology, University of Sassari,
Italy
The effects of microgravity on T lymphocytes was studied
on three sounding rockets flights with µg phases lasting 7-
13 min. Automated pre-programmed instruments could inject
fluorescent-labelled concanavalin A and/or para-formaldehyde as
fixative at given times. A microscope remotely controlled from
the ground and connected to a CCD camera recorded cell motility
and interactions under µg. Chemically fixed cells were
labelled after flight with monoclonal anti-vimentin and anti-
tubulin, respectively, followed by fluorescent anti-mouse Ig.
First, while binding of concanavalin A to the cell membrane is
not affected, patching of the receptors is slightly but
significantly retarded during µg. Second, marked alterations
of the intermediate filaments of vimentin as well as of the
microtubule network are observed after 30 s of µg and change
only slightly during the flight. Ground controls conducted at
hypergravity show that the changes are not due to the
acceleration peak of 13-15 g following launch. Third, cell
motility and cell-cell contacts also occur when cells are free-
floating in µg. These results indicate that direct effects
of µg on the cytoskeleton are likely whereas little changes
occur at the membrane level, and are in agreement with recent
data showing that gravity controls the pattern of microtubule
formation. The existence of active movements and cell-cell
contacts supports, but does not prove, the notion that T cells
can communicate and transmit signals in µg.
Introduction
A dramatic depression of T lymphocyte activation in vitro by
concanavalin A (Con A) was discovered in an experiment conducted
aboard Spacelab 1 in 1983.¹ More sophisticated experiments
followed during Spacelab D1 in 1985,² SLS-1 in
1991³,4 and IML-2 in 1994.5 All
investigations confirmed the effect. The results indicate that
the depression is mainly due to a lack of expression of the
interleukin-2 receptor.4, 5 Conversely, when T
lymphocytes and monocytes were attached to microcarrier beads,
activation was more than doubled under µg compared with
controls cultured at 1 g, either in flight or on the
ground.3, 4 It was concluded that signal transduction
is markedly changed in µg.
The work described in this paper was carried out to clarify
aspects of such effects that can be investigated during
relatively short µg periods (7-13 min) achieved on sounding
rockets using either automatic pre-programmed instruments or a
microscope adjusted via telecommand.
The first question addressed was whether binding of Con A to
the cell membrane, followed by patching and capping of the
membrane proteins involved, would take place normally in µg.
It is believed that this is the first signal required for
activation.6 Patching and capping are processes
occurring within a few minutes after binding. Abnormalities in
this process would obviously alter the transduction of the first
activation signal. The hypothesis was that exposure to µg
could produce changes of certain membrane properties.
The second question was related to structural changes possibly
occurring within the cells exposed to µg. Gravity unloading
per se is not sufficient to change the shape of a cell, because
the microenvironment is subjected to forces that are many orders
of magnitude larger than that of gravity.7, 8 However,
organelles with a higher density than the cytoplasm (such as
ribosomes, centrioles and nucleolus) may exert at 1 g some
pressure on cytoskeletal and other structures. This pressure
would disappear in µg. A similar effect occurs in the
statocytes of the roots of some plants where gravitropism is
attributed to the pressure of the statoliths on the endoplasmic
reticulum. Structural changes may have an important impact on
cellular events. In addition, their occurrence would demonstrate
the existence of direct effects of µg on single mammalian
cells.
The third question was whether free-floating cells in µg
are capable of autonomous motions and, hence, of cell-cell
interactions. Cell-cell interactions are an important means of
cell communication and signal delivery in T lymphocyte
activation. Although in previous experiments cell aggregates were
always observed in samples fixed in µg,¹,² one
could argue that lack or reduction of cell contacts is the cause
of the loss of activity in space. Direct evidence in real time
will certainly give a clear answer to the question.
We present here the results of four experiments carried out
on the MASER 3, MASER 4 and MAXUS 1B sounding rockets. In MASER
3/4, the binding of fluorescent-labelled Con A (FITC-Con A) to
human lymphocytes and the subsequent patching and capping was
investigated. In MAXUS 1B, one experiment studied the structure
of the microfilaments of vimentin and of the microtubule network
in Jurkat cells (a human T cell line) chemically fixed in-flight
and labelled after recovery with monoclonal antibodies. In the
second experiment, motility and interactions of human lymphocytes
were recorded in real time by an onboard microscope. A detailed
description of the instruments aboard MASER 3/4 is given in ref.
9, while the equipment used on MAXUS is presented in ref. (6).
Materials and methods
Flight instruments
All cell cultures were
incubated at 37°C during flight. In MASER 3, the experiment
was installed in the CIS-1 (Cells in Space) module developed by
Fokker Space & Systems with the National Aerospace Laboratory and
the Centrum voor Constructie en Mechatronica (CCM) in The
Netherlands. The experiment-specific device consisted of a unit
provided by CCM. Cell cultures and reagents were stored in three
contiguous sections (respectively, fixative/cells/Con A) of a
flexible silicon tube closed at both ends and separated by roller
bars released at pre-programmed times. See ref. 9 and Section 3
of this volume for details.
In MASER 4, a more sophisticated system, the Plunger Box Mix
Unit (PBMU, also provided by CCM), was used in the CIS-2 module.
The reagents and, later, the fixative were injected into the
cultures by pre-programmed plungers.9
Two instruments, both integrated in the TEM 06-5M module and
provided by ERNO Raumfahrttechnik, Bremen, were used in MAXUS 1B
(see ref. 6 for a detailed description). To study the
cytoskeleton, cell cultures and fixative were sealed in two
disposable syringes separated by a valve. At pre-programmed
times, the piston of the syringe with the fixative automatically
injected the reagent into the culture. The observation of cell
movements was performed in a glass cuvette mounted in a phase
contrast microscope connected to a CCD camera. To prevent
sedimentation of the cells, the cuvette was rotated at 30 rpm/min
during the pre-flight and launch phases; rotation was stopped at
the onset of µg. In-flight, the selection of the observation
field and focusing were operated manually via telecommand from
the ground station.
Each flight experiment with the exception of the observation
of cell movements was accompanied by a synchronous ground
control with the same batch of cells in an equivalent apparatus.
Human lymphocytes
Peripheral blood was
drawn from healthy donors 14-18 hr before launch. Cells were
purified by gradient centrifugation on Ficoll (Pharmacia,
Uppsala) and cultures were prepared as described
previously.10 The preparations contained 84±3%
of lymphocytes. The remaining were macrophages (13±3%) and
polymorphonuclear cells.
Jurkat cells
Jurkat cells, derived from
a human T cell leukemia,11 were kindly provided by A.
Lanzavecchia of the Basle Institute of Immunology, Basle. The
cells are mycoplasma-free. Cells were grown in RPMI 1640 medium
supplemented with 10% (v/v) foetal bovine serum (Biochrom KG-
Germany), 20 mM HEPES, 5 mM sodium bicarbonate, 1 mM sodium
pyruvate, 4 mM L-glutamine and gentamycine (50 µg/ml).
Markers
Monoclonal antibodies anti-
vimentin (V-5255) and anti-ß tubulin (T-4026), and anti-
mouse polyvalent immunoglobulins FITC-conjugated (fluorescein
isothiocyanate) were obtained from SIGMA (St. Louis, Missouri,
USA). Con A-FITC was purchased from ICN Immuno Biologicals
(Lisle, Illinois, USA).
Flight investigations
In all
investigations, the experiment-specific units containing the cell
cultures and reagents were installed in the modules of the
rockets 2 hr before launch. The µg phases of MASER 3 & 4
lasted 7 min, that of MAXUS 1B 12.5 min. µg in a sounding
rocket flight is defined as g <10-4. The flight
samples were recovered and handed over to the experimenters
within 90 min of launch.
In MASER 3, lymphocyte cultures (290 µl, 2x106
cells/ml) were sealed in the middle part of each of the eight
silicon tubes of the flexible tube unit. After onset of µg,
FITC-Con A (280 µl, 51 µg/ml, final concentration 25
µg/ml) was added. Fixation followed by mixing with
paraformaldehyde (final concentration 1%) 0.3, 30.3, 60.3, 90.3,
120.3, 180.3, 270.3 & 390 s after the addition of FITC-Con A. The
protocol of the investigation in MASER 4 was analogous to that
of MASER 3. In four of the six chambers, FITC-Con A was released
180 s after onset of µg; fixations followed 0.3, 30, 90 &
210 s later. In the other two chambers the mitogen was added
after 300 s of µg; fixations followed 30 & 90 s later. All
samples were duplicated.
In MAXUS 1B, the labelling of vimentin was carried out on
Jurkat cells fixed 30, 420 & 720 s after the onset of µg by
means of an automatic pre-programmed device. For tubulin
labelling, the cells were fixed after 30 & 720 s. To 1 ml of
culture of Jurkat cells (1.7x106 cells/ml), 1 ml of
2% paraformaldehyde was added. The observation of cell motility
was made in cultures containing 3x106 lymphocytes/ml.
Realtime recording was on magnetic tape in an onboard CCD camera.
Analysis was performed on a papercopy (video-printout) showing
pictures taken at 13 s intervals.
Evaluation of binding, patching and capping
After recovery, the cells were washed three times
with Hanks balanced salt solution (PBS) and fixed on microscope
slides by cytocentrifugation (200 g, 8 min). The cells were
mounted in glycerol containing p-phenylendiamine as antifading
and observed in a Nikon Optiphot microscope. At least three
slides were prepared from each sample. About 500 cells were
evaluated and counted on each slide. For statistical analysis of
the data the one-tailed U-test of Wilcoxon, Mann and Whitney was
applied.
Labelling of vimentin and tubulin
After
recovery, the cells were rinsed with 10 ml of cytoskeleton-
stabilising buffer (SB) containing 50 mM imidazole, 50 mM
potassium chloride, 0.5 mM magnesium chloride, 1 mM EGTA, 1 mM
2-mercaptoethanol, 4 M glycerol, pH 6.8. The cell pellet was
resuspended in 1 ml of SB containing 1% Triton X-100 and the
cells were kept for 30 min at ambient temperature. The cells were
rinsed again with 10 ml SB, pelleted and incubated for 30 min at
ambient temperature with 200 µl of anti-vimentin or anti-
tubulin, diluted 1:100 with SB. At the end of the incubation the
cells were rinsed with 10 ml of SB, and the cell pellet incubated
with the secondary antibody FITC-conjugated, diluted 1:100 in SB.
Incubation and rinsing with SB followed as above. Finally, the
cells were cytocentrifuged (200 g, 8 min). The cells, adhering
to microscope slides, were mounted in glycerol containing p-
phenylendiamine as antifading and observed in epifluorescence in
a Nikon Optiphot microscope. Three slides were prepared from each
sample and about 400 cells were evaluated on each slide.
Micrographs were made on Fujichrome 400 ASA films. For
statistical analysis of the data the one-tailed U-test of
Wilcoxon, Mann and Whitney was applied.
Results and discussion
Binding of the mitogen
The type and
number of signals required for T lymphocyte activation have yet
to be clarified and are still a matter of
controversy.6 Nevertheless, there is agreement in the
literature on the nature of the first signal. This is usually
provided by the presentation to the T cell of the modified
antigen in conjunction with the MHC component of the antigen-
presenting cell, usually a monocyte. The presentation of the
antigen can be replaced by a mitogen such as Con A. At least part
of the effects observed in space could be attributed to
alterations of the binding pattern. Therefore, it was essential
to establish whether the binding of Con A to the cell membrane,
followed by patching and capping of the receptors involved,
occurs normally in µg.
Two experiments were conducted to answer this question;
preliminary data were presented in conference
proceedings.12, 13 In the first
investigation (MASER 3), Con A was added to the cells immediately
after µg conditions were established. Fixation with
paraformaldehyde followed after different preset times of 0.3-390
s. No significant differences in the rate of binding, patching
and capping compared to the 1 g control were
observed.12 Binding was very fast and completed after
30 s.
The second experiment (MASER 4) was carried out to test if the
lack of differences between flight and 1 g control samples arose
from a lack of influence of µg or from the fact that the
cells did not have sufficient exposure time to 'adapt' to
µg. Therefore, Con A was injected into the samples 3 & 5 min
after the onset of µg, followed by fixation after different
preset times. The data obtained confirm that, even after 5 min
in µg, there are no changes in binding of Con A to the cell
membrane compared to the ground control.13
Furthermore, there are no differences in the patching of the Con
A receptors in the samples where the cells have been exposed for
3 min to µg before the addition of the mitogen (Fig. 1).
However, patching is significantly retarded when Con A is added
after 5 min of µg (Fig. 1), especially in the samples where
the cells were fixed 90 s after the addition of Con A. In the
flight samples, 2.9±0.9% of the cells show patching,
compared to 7.4±3.4% in the 1 g control (99.5% significance
of the difference; U(11.6) = 5, alpha = 0.005). In the samples
fixed after 30 s, the difference in patching is statistically
significant at the 95% level (1.9±1.0% in the flight sample,
compared to 3.9±2.6% in the ground control; U(12,7) = 19,
alpha = 0.05). Furthermore, we also observed a significant
difference in patching between the samples exposed for 3 or 5 min
to µg and fixed after 90 s (97.5% significance level; U(6,6)
= 3, alpha = 0.025). In addition, the data in Fig. 1 indicate
that, despite a significantly lower rate at µg, patching is
a dynamic process that would be completed if the incubation time
were longer than 210 s. The question on differences in capping
could not be answered with statistically significant data as the
number of cells showing capping was too low in this experiment.
However, there seems to be a tendency that capping is retarded
in the flight samples.
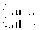
Fig. 1. Patching and capping of Con A receptors following
binding of the mitogen to human lymphocytes. The bars represent
the percentage of cells showing either patching or capping at
different fixation times after the addition of Con A. Standard
errors of the means are given.
The fact that the patterns of binding, patching and capping
are not changed compared to the ground control when Con A is
added immediately after the onset of µg, and the fact that
changes in patching - but not in binding - are observed only when
Con A is added after 5 min of µg, indicate that the short
exposure to hypergravity during the launch phase is not relevant.
The data show that the influence of µg on the delivery
of the first activation signal is rather small and that,
therefore, rapid processes such as binding of the mitogen,
patching (although slightly retarded) and probably also capping
are not involved in the depression of the in vitro activation of
T lymphocytes observed in the Spacelab experiments.1-5
Cytoskeleton
In connection with the study
on cell movements in µg, we investigated the occurrence of
cytoskeleton changes as it is well known that the cytoskeleton
plays an important role in cell motility. In particular, we
analysed the pattern of the intermediate filaments of vimentin
and of microtubuli in Jurkat cells. The higher cytoplasmic volume
and size of these cells, compared to resting lymphocytes, render
them more suitable for this type of analysis.
The cells were fixed with paraformaldehyde 30, 420 & 720 s
after the onset of µg. After recovery, the cytoskeleton was
marked with monoclonal antibodies anti-vimentin and anti-tubulin.
The cytofluorographs of the intermediate filaments of vimentin
are shown in Fig. 2; the quantitative evaluation of the changes
is presented in Fig. 3.

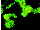
Fig. 2. Cytofluorographs of the intermediate filaments of
vimentin of Jurkat cells. Top: ground control; Bottom: flight
sample fixed after 30 s in µg.
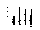
Fig. 3. Influence of gravity changes on the structure of Vimentin in Jurkat cells. The bars represent
the percentage of cells showing either the appearance of large bundles (solid bars) or the formation
of protein aggregates together with discontinuities of filamentous network (cross-hatched bars).
Standard errors of the means are given.
The morphological analysis reveals that cytoskeleton
structural changes are occurring in µg and that they are
most evident in vimentin. We must point out that some of the
changes were also detected, although to a significantly smaller
degree, in the control samples.
The most important difference is given by the intermediate
filaments of vimentin which under µg assemble into thick
bundles in 20.9±1.7% of the cells, compared to 9.9±1.3%
(95% significance of the difference; U(3,3) = 0, alpha = 0.05)
in 1 g. This change is observed after only 30 s of µg. The
number of cells showing large bundles is slightly but
significantly decreased at µg times of 420 s
(17.8±0.4%, 95% significance, U(3,3) = 0, alpha = 0.05) and
720 s (18.8±1.6%, 95% significance, U(3,3) = 0, alpha =
0.05).
The other changes observed in µg consist of the formation
of aggregates of protein, appearing as fluorescent points in Fig. 2
(bottom) and suggesting depolimerisation processes, as well as
of discontinuities of the filamentous network (11.9±1.3% at
30 s, 12.1±2.3% at 420 s and 11.0±1.3% at 720 s,
compared to 8.8±1.6% of the ground control. Also, these
changes are significant (95% at 30 s and 90% at 420 & 720 s).
A significant difference in the appearance of large bundles
has also been observed in the microtubuli network, where
13.2±1.2% of the cells show these changes after 30 s of
µg, compared to 5.9±2.8% in the ground control (95%
significance, U(3,3) = 0, alpha = 0.05). After 720 s,
11.8±1.5% of the cells show large bundles. Also, this
difference is significant (95%, U(3,3) = 0, alpha = 0.05). The
other changes are less evident.
During the sounding rocket's boost phase, the cells were
exposed to hypergravitational forces. In a control experiment,
cells loaded into flight-type syringes were exposed for 2 min to
13 g at 37°C and than fixed. Analysis of the structure of
vimentin showed the appearance of large bundles in 8.9±1.2%
of the cells, whereas the formation of aggregates was found in
8.0±1.1%. Thus these cells do not exhibit the effects
observed in µg. The same is true for the microtubuli network
where 7.55±1.2% of the cells exposed to hypergravity showed
large bundles. Therefore, the observed changes in the
cytoskeletal structure are due to µg and not to the g-stress
caused by the launch or a combination of hypergravity and
gravitational unloading. This is also supported by the fact that
the number of cells displaying the changes are only slightly
reduced after 720 s in µg.
The intermediate filaments of vimentin consist of a protein
expressed in cells of mesenchimal origin as well as in other
types of cells cultured in vitro.14 The proposed
function of the intermediate filaments is to provide a structural
network, with characteristics typical of each cell type. This
network maintains the shape of the cell, the distribution of its
organelles and, based on the connections between cytoplasmic
membrane and nucleus, mediates the intracellular signal
transduction.
Little is known on the function of vimentin in lymphocytes:
Studies carried out on B cells with anti-immunoglobulins
antibodies, have demonstrated that this protein is involved in
the capping of surface immunoglobulins in the region of the
uropodes.15
Of particular interest is the observation that in T
lymphocytes, the activation by Con A causes an alteration in the
distribution of vimentin that appears to be correlated with the
phases of the cellular cycle.16 This indicates that
the role of vimentin may change during the progression of the
cell through its cycle. In addition, cytoskeleton changes, beside
generating the movements, may reveal the beginning of the
apoptosis which was observed in micrographs of lymphocytes
cultivated in an earlier experiment in space²
A recent article 17 discussed the role of gravity
in pattern formation of microtubule in solutions of purified
tubulin. The pattern was different depending on whether the
reaction containers were in the upright or horizontal position.
This phenomenon is related to the bifurcations occurring in non-
linear out of equilibrium states. As discussed by the authors in
a previous paper,18 the transition from 1 g to µg
may result in a bifurcation point at which the biological systems
drastically changes its behaviour.
The data presented here, together with the authors' previous
findings in space,1-5 support the bifurcation theory
and the control of pattern formation of cytoskeletal structures
by gravity.
Cell motility
The images recorded under
µg clearly show that the free-floating non-activated cells
were able to display autonomous motion in random directions. Fig. 4A
shows the overall displacement of 11 different cells. A
detailed analysis (one image every 13 s) shows that the movements
were much more complex. The cells often changed direction, moved
back and forwards and sometimes crossed the same point several
times (Fig. 4B). The average velocity, calculated from the
displacement in the 13 s increment, was 8.4±1.2 µm/min,
with a range of 0-29.4 µm/min. Also of interest is the
observation that the cells in µg were not all round. They
very often exhibited longitudinal forms, rotated around their
axis and also showed contraction waves similar to those described
in the literature for lymphocytes that move in collagen gels
under 1 g conditions.19 All 11 cells in the
observation field showed motion capability under µg
conditions. This is in contradiction to the behaviour of
lymphocytes under 1 g conditions, where mostly activated cells
only or cells in the presence of a chemoattractant show this
capability.20
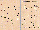
Fig. 4. Motility and interactions of human lymphocytes in
µg. A: overall movement of 11 different cells; B: detailed
motion behaviour of four cells (one measuring point each 13 s).
The numbers on the axes, in µm, correspond to the position
of the cell in the viewing field.
The origin of the movement can be attributed to two major
causes. First, changes of the cytoskeleton, in particular the
formation of thick bundles of microfilaments, may induce motility
of lymphocytes by contraction and elongation of the free-floating
cell. Second, the Marangoni convection due to differences in the
concentration of components dissolved in the medium may generate
the movements of cells resuspended in µg.21,
22Concentration gradients are generated by the metabolism
of the cells, which are consuming nutrients (mainly glucose and
glutamine) and producing waste (such as lactate and ammonia).
Recent data (Cogoli-Greuter, Dall'Aglio, Bedarida and Cogoli,
unpublished results) based on laser diffraction studies clearly
show that concentration gradients form in cultures of lymphocytes
and Jurkat cells.
In a recent experiment performed in the NIZEMI unit during the
Space Shuttle's IML-2 mission, the motility and aggregate
formation of human lymphocytes during the in vitro activation
with Con A was observed in real time.23 The velocity
of free cells (cells not attached to an aggregate) in the
presence of Con A was found to be in the same range as in the
absence of the mitogen: 6.6±1.4 µm/min. Interestingly,
the velocity did not change during the 3 days of incubation,
whereas in the ground control, the velocity of single cells was
significantly lower and decreased with increasing incubation
time.
Nevertheless, although the movements and interactions observed
under µg in free-floating cells suggest that cell-cell
interactions and signal transmission may work as at 1 g, it is
important to remark that the cytoskeleton is altered and that,
therefore, signal transmission and transduction may be altered
as well. Also, the aggregates formed by lymphocytes are smaller
and less frequent under µg than at 1 g 2, 23
Conclusions
The experiments described here have contributed to clarifying
certain aspects of the in vitro activation of T lymphocytes in
microgravity:
-
The reception of the first signal required for activation, namely
the binding of Con A to alpha-glucosides of membrane proteins,
is not affected by µg, whereas patching and probably also
capping are slightly retarded. This indicates that neither the
structure and function of the cell membrane nor the interaction
receptor/ligand are altered by µg. Similar conclusions were
drawn from experiments conducted on parabolic flights with gap
junctions extracted from cardiac tissue,24 with
rhyzobia and lectin,25 and with alpha-feto protein and
monoclonal antibodies.26
- The changes of the microfilaments and microtubuli
patterns suggest that single cells in culture may experience
direct effects of gravity unloading. Whether this contributes to
the alterations of the proliferation rate and of the genetic
expression observed in the in vitro activation of T cells in
long-duration space flight has to be assessed in further
experiments. However, the detection of direct effects of gravity
is one of the crucial tasks of gravitational biology. Indirect
effects, conversely, are attributable to changes of the
microenvironment due to the lack of convection and sedimentation.
- For the fist time, motion of mammalian cells in µg
was observed and analysed at the microscope. The cell movements
can be attributed to the thermosolutal Marangoni convection and
to cytoskeletal changes. Again, further experiments have to
clarify the question whether the contacts seen under µg are
sufficiently tight to permit the transmission of signals between
the cells.
Acknowledgements
This project was conducted with grants from the Italian Space
Agency (ASI), the Swiss National Research Foundation (Grant No.
31-25181.88), ETH Zürich, Contraves A.G., Zürich, and
ERNO Raumfahrttechnik, Bremen. The authors gratefully acknowledge
the assistance of D. Mesland, W. Herfs and D. Frimout of ESA-
ESTEC, Noordwijk, of R. Huijser of Fokker Space & Systems,
Amsterdam, of H. van Soest and A. Koppen of CCM, Nuenen (NL), of
the team of ERNO Raumfahrttechnik, Bremen, in particular D.
Grothe and I. Meyer, and of the team of the Swedish Space
Corporation at the ESRANGE launch facility in Kiruna, Sweden. The
authors also wish to thank A. Lanzavecchia of the Basle Institute
of Immunology for providing the Jurkat cells.
References
-
Cogoli, A., Tschopp, A. & Fuchs-Bislin, P. (1984). Cell
sensitivity to gravity. Science 225, 228-230.
-
Cogoli, A., Bechler, B., Müller, O. & Hunzinger, E. (1988).
Effect of microgravity on lymphocyte activation. In Biorack
on Spacelab D1 (eds. Longdon, N. & David, V.). ESA SP-1091,
89-100.
-
Bechler, B., Cogoli, A., Cogoli-Greuter, M., Müller, O. &
Hunzinger, E. (1992). Activation of microcarrier-attached
lymphocytes in microgravity. Biotechnol. Bioeng. 40, 991-
996.
-
Cogoli, A., Bechler, B., Cogoli-Greuter, M., Criswell, S. B.,
Joller, H., Joller, P., Hunzinger, E. & Müller, O. (1993).
Mitogenic signal transduction in T lymphocytes in microgravity.
J. Leukoc. Biol. 53, 569-575.
-
Pippia, P., Sciola, L., Cogoli-Greuter, M., Meloni, M. A., Spano,
A. & Cogoli, A. (1996). Activation signals of T lymphocytes in
microgravity. J. Biotechnol. 47, 215-222.
-
Cogoli, A. (1993). The effect of hypogravity and hypergravity on
cells of the immune system. J. Leukoc. Biol. 54, 259-268.
-
Albrecht-Buehler, G. (1990). In defense of 'non-molecular' cell
biology. Int. Rev. Cytol. 120, 191-241.
-
Albrecht-Buehler, G. (1991). Possible mechanisms of indirect
gravity sensing by cells. ASGSB Bulletin 4, 25-39.
-
Huijser, R., Aartman, L. & Willemsen, H. (1990). Cells in space:
sounding rocket facilities for cell biology and biotechnology in
microgravity. In Proc. 4th European Symp. Life Sciences Res.
in Space, Trieste (ed. David, V.). ESA SP-307, 455-466.
-
Tschopp, A. & Cogoli, A. (1983). Hypergravity promotes cell
proliferation. Experientia 39, 1323-1329.
-
Weiss, A., Wiskocil, R. & Stobo, J. (1984). The role of T3
surface molecules in the activation of human T cells: a two-
stimulus requirement for IL 2 production reflects events
occurring at a pre-translational level. J. Immunol. 133,
123-128.
-
Cogoli, M., Bechler, B., Cogoli, A., Arena, N., Barni, S.,
Pippia, P., Sechi, G., Valora, N. & Monti, R. (1990). Lymphocytes
on sounding rockets. In Proc. 4th European Symp. Life Sciences
Res. in Space, Trieste (ed. David, V.). ESA SP-307, 229-234.
-
Cogoli, M., Bechler, B., Cogoli, A., Arena, N., Barni, S.,
Pippia, P., Sechi, G., Valora, N. & Monti, R. (1992). Lymphocytes
on sounding rockets. Adv. Space Res. 12, 141-144.
-
Lazarides, E. (1980). Intermediate filaments as mechanical
integrators of cellular space. Nature 283, 249-256.
-
Dellagi, K. & Brouet, J. C. (1982). Redistribution of
intermediate filaments during capping of lymphocyte surface
molecules. Nature 298, 284-286.
-
Paulin-Levasseur, M. & Brown, D. L. (1987). Vimentin dynamics
during the mitogenic stimulation of mouse splenic lymphocytes.
Cell Motil. Cytoskeleton 8, 227-237.
-
Tabony, J. (1994). Morphological bifurcations involving reaction-
diffusion processes during microtubule formation. Science
264, 245-248.
-
Cogoli, A. & Gmünder F. K. (1991). Gravity effects on single
cells: Techniques, findings and theory. In Advanced Space
Biology and Medicine (ed. Bonting S. L.). Vol I, 193-248, JAI
Press Inc.
-
Haston, W. S. & Shields, J. M. (1984). Contraction waves in
lymphocyte locomotion. J. Cell Sci. 68, 227-241.
-
Wilkinson, P. C. (1987). Leukocyte locomotion: Behavioural
mechanisms for accumulation. J. Cell Sci. Suppl. 8, 104-
119.
-
Scriven, L. E. & Sternling, C. V. (1960). The Marangoni effects.
Nature 187, 186-188.
-
Napolitano, L. G. (1984). Marangoni convection in space
microgravity experiments. Science 225, 197-198.
-
Cogoli-Greuter, M., Meloni, M. A., Sciola, L., Spano, A., Pippia,
P., Monaco, G. & Cogoli, A. (1996). Movements and interactions
of leukocytes in microgravity. J. Biotechnol. 47, 279-287.
-
Claassen, D. E. & Spooner, B. S. (1989). Effects of microgravity
on liposome reconstituted cardiac gap junction channeling
activity. Biochem. Biophys. Res. Commun. 161, 358-63.
-
Henry, R. L., Green, P. D., Wong, P. P. & Guikema, J.A. (1990).
Binding of isolated plant lectin by rhizobia during episodes of
reduced gravity obtained by parabolic flight. Plant Physiol.
92, 262-64.
-
Spooner, B. S., Guikema, J. A. & Barnes, G. (1990). Binding of
alpha fetoprotein by immobilized monoclonal antibodies during
episodes of zero-gravity obtained by parabolic flight. Aviat.,
Space & Environ. Med. 61, 725-28.
About|
Search|
Feedback



 SP1206
SP1206
Published April 1997.
Developed by ESA-ESRIN ID/D.




